学习目标了解计数数据变换方法的重要性了解 PCA (principal component analysis)了解如何使用 PCA 和层次聚类评估样本质量1. 质控
DESeq2 工作流程的下一步是 QC,其中包括样本和基因程度上,以对计数数据执行 QC 检查,以帮助我们确保样本或重复看起来良好。
QC
2. 样本QC
RNA-seq 分析中一个有用的初始步骤通常是评估样本之间的整体相似性:
哪些样本彼此相似,哪些不同?这是否符合实验设计的预期?数据集中的主要变异来源是什么?
为了探索样本的相似性,我们将使用主成分分析 (PCA) 和层次聚类方法执行样本级 QC。这些方法或工具使我们能够检查重复彼此之间的相似程度(聚类),并确保实验条件是数据变化的主要来源。样品级 QC 还可以帮助识别任何表现出异常值的样品;我们可以进一步探索任何潜在的异常值,以确定是否需要在 DE 分析之前将其删除。
Sample-level QC
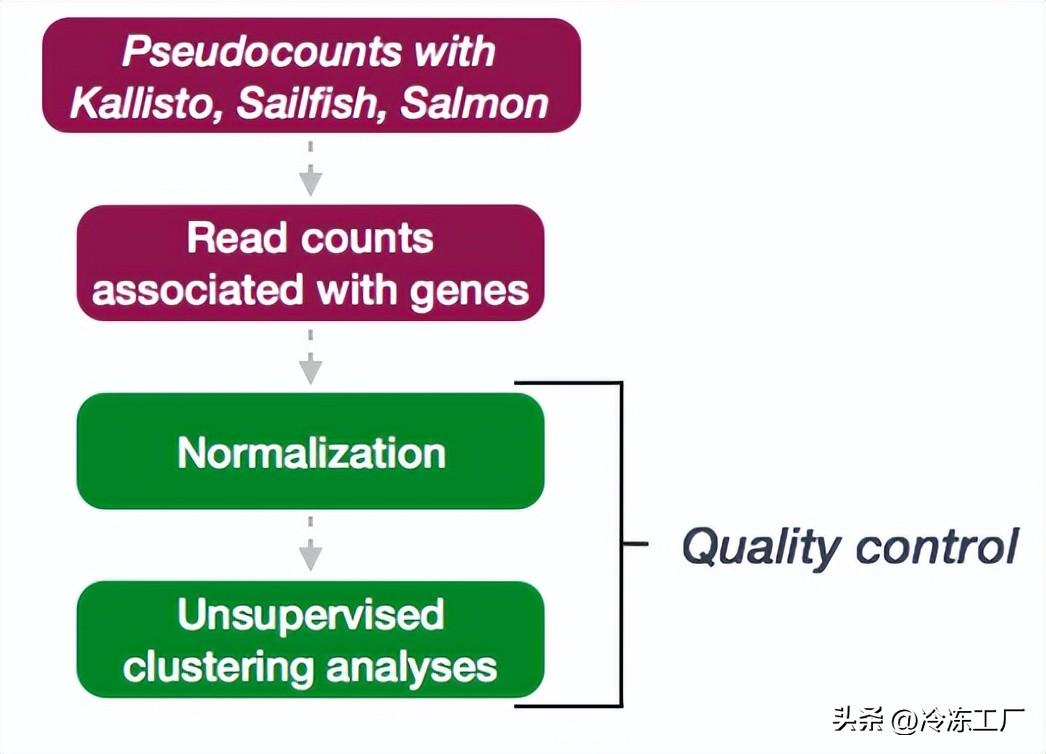
这些无监督聚类方法使用 log2 变换的归一化计数运行。log2 转换改进了可视化的距离。我们将不使用普通的 log2 变换,而是使用正则化对数变换 (rlog),以避免因大量低计数基因而产生的任何偏差;
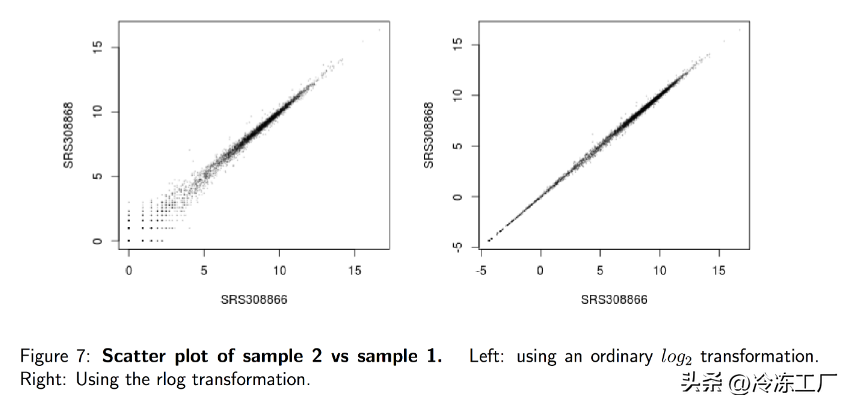
transformation
许多用于多维数据探索性分析的常用统计方法,尤其是聚类和排序方法(例如,主成分分析等),最适合(至少近似地)同方差数据;这意味着可观察量的方差(即,这里是基因的表达值)不依赖于均值。然而,在 RNA-seq 数据中,方差随平均值增加。例如,如果直接对归一化读取计数矩阵执行 PCA,则结果通常仅取决于少数高表达的基因,因为它们在样本之间显示出最大的绝对差异。避免这种情况的一种简单且经常使用的策略是「取归一化计数值的对数加上一个小的伪计数」;然而,现在具有低计数的基因往往主导结果,因为由于小计数值固有的强泊松噪声,它们在样本之间显示出最强的相对差异。
因此,DESeq2 提供了「正则化对数转换」,或简称为 rlog。对于计数高的基因,rlog 转换与普通的 log2 转换差别不大。然而,「对于计数较低的基因,这些值会缩小到所有样本中基因的平均值」。这样做是为了使 rlog 转换后的数据近似同方差。
❝
DESeq2 建议大型数据集(100 个样本)使用方差稳定变换 (vst) 而不是 rlog 来进行计数变换,因为 rlog 函数可能需要运行很长时间,而 vst() 函数在类似情况下更快。
❞
3. PCA
主成分分析 (PCA) 是一种用于强调变化并在数据集中降维的技术。这是一种非常重要的技术,用于质量控制和 Bulk RNA-seq 和单细胞 RNA-seq 数据的分析。
3.1. PCA plots
本质上,如果两个样本的基因表达水平相似,这些基因对给定 PC(主成分)表示的变异有显著贡献,则它们将在表示该 PC 的轴上靠近绘制。因此,我们期望生物重复具有相似的分数(因为我们的期望是相同的基因正在发生变化)并聚集在一起。通过可视化一些示例 PCA 图最容易理解这一点。
我们在下面有一个示例数据集和一些相关的 PCA 图,以了解如何解释它们。实验的元数据如下所示。感兴趣的主要条件是处理。
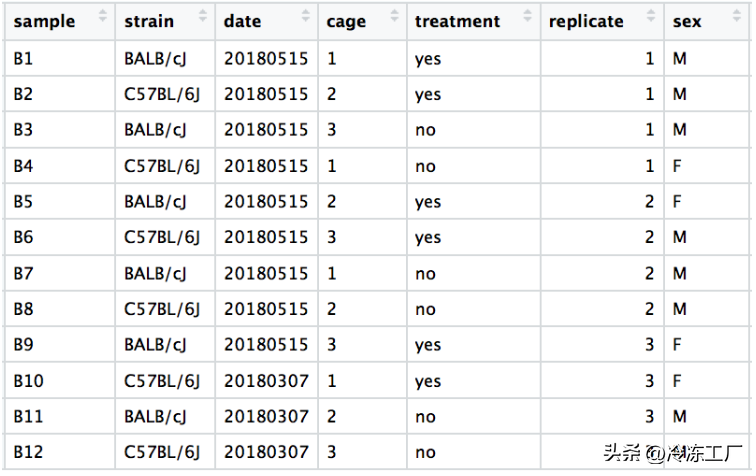
dataset
在 PC1 和 PC2 上进行可视化时,我们没有看到样本因处理而分开,因此我们决定探索数据中存在的其他变异来源。我们希望我们已经在我们的元数据表中包含了所有可能的已知变异源,并且我们可以使用这些因素来为 PCA 图着色。
PCA_1
我们从cage因子开始,但cage因子似乎无法解释 PC1 或 PC2 上的变化。
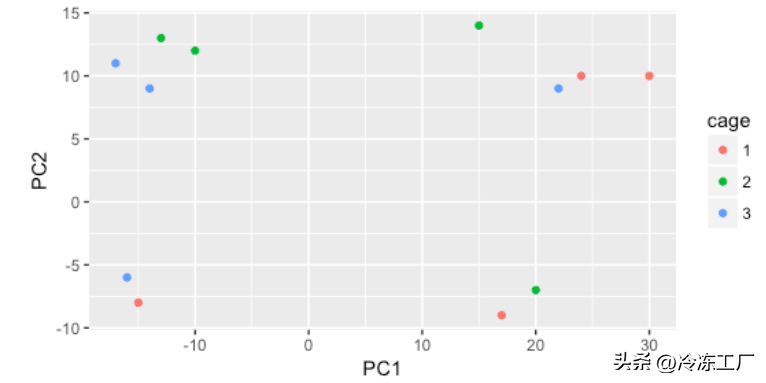
cage
然后,我们按 sex 因素着色,这似乎在 PC2 上分离样本。这是需要注意的好信息,因为我们可以在下游使用它来解释模型中由于 sex 引起的变化并将其回归。

sex
接下来我们探索 strain 因子,发现它可以解释 PC1 上的变化。
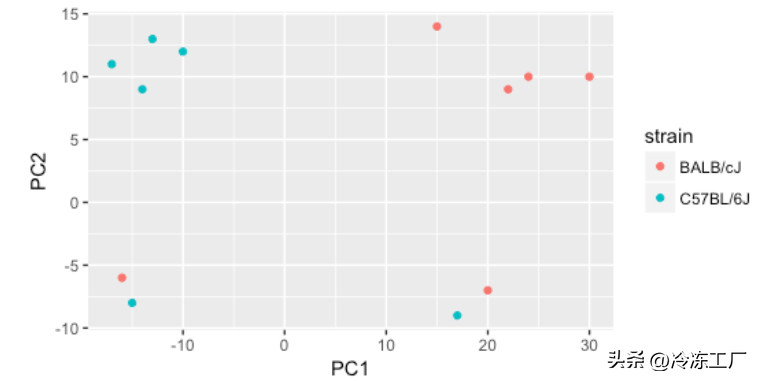
strain
很高兴我们能够确定 PC1 和 PC2 的变异来源。通过在我们的模型中考虑它,我们应该能够检测到更多因处理而差异表达的基因。
令人担忧的是,我们看到两个样本没有与正确的 strain 聚类。这表明可能存在样本交换,应进行调查以确定这些样本是否确实是标记的 strain。如果我们发现有一个交换,我们可以交换元数据中的样本。但是,如果我们认为它们被正确标记或不确定,我们可以从数据集中删除样本。
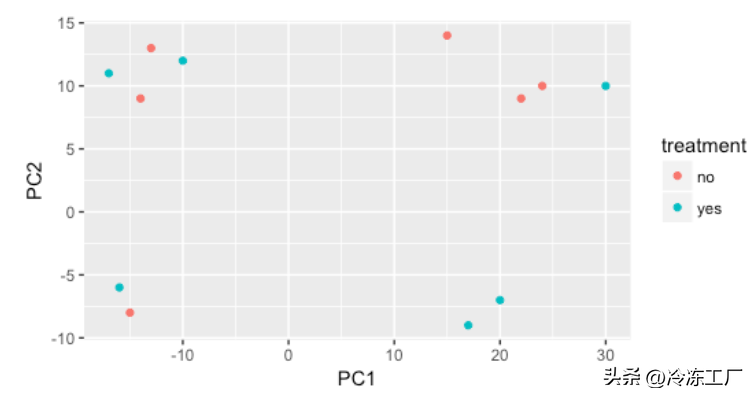
我们仍然没有发现处理是否是 strain 和 sex 后变异的主要来源。因此,我们探索 PC3 和 PC4 以查看处理是否正在推动由这两个 PC 中的任何一个代表的变异。
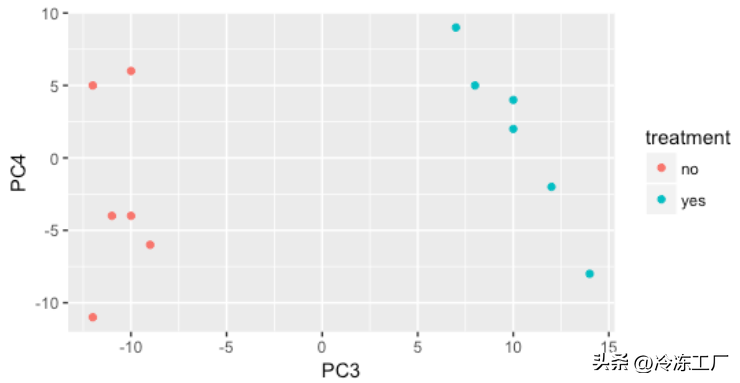
treatment
我们发现样本在 PC3 上通过处理分离,并且对我们的 DE 分析持乐观态度,因为我们感兴趣的条件,处理,在 PC3 上分离,我们可以回归驱动 PC1 和 PC2 的变化。
根据前几个主要成分解释了多少变化,您可能想要探索更多(即考虑更多成分并绘制成对组合)。即使您的样品没有通过实验变量清楚地分离,您仍然可以从 DE 分析中获得生物学相关的结果。如果您期望效果大小非常小,那么信号可能会被无关的变异源淹没。在您可以识别这些来源的情况下,在您的模型中考虑这些来源很重要,因为它为检测 DE 基因的工具提供了更多功能。
4. 层次聚类
与 PCA 类似,层次聚类是另一种互补的方法,用于识别数据集中的模式和潜在异常值。热图显示数据集中所有成对样本组合的基因表达相关性。由于大多数基因没有差异表达,样本之间通常具有很高的相关性(值高于 0.80)。低于 0.80 的样本可能表示您的数据和/或样本污染中存在异常值。
沿轴的分层树指示哪些样本彼此更相似,即聚集在一起。顶部的色块表示数据中的子结构,您会希望看到您的重复一起作为每个样本组的一个块。我们的期望是样本聚集在一起类似于我们在 PCA 图中观察到的分组。
在下图中, Wt_3 和 KO_3 样本没有与其他重复聚类在一起。我们想要探索 PCA 以查看我们是否看到相同的样本聚类。
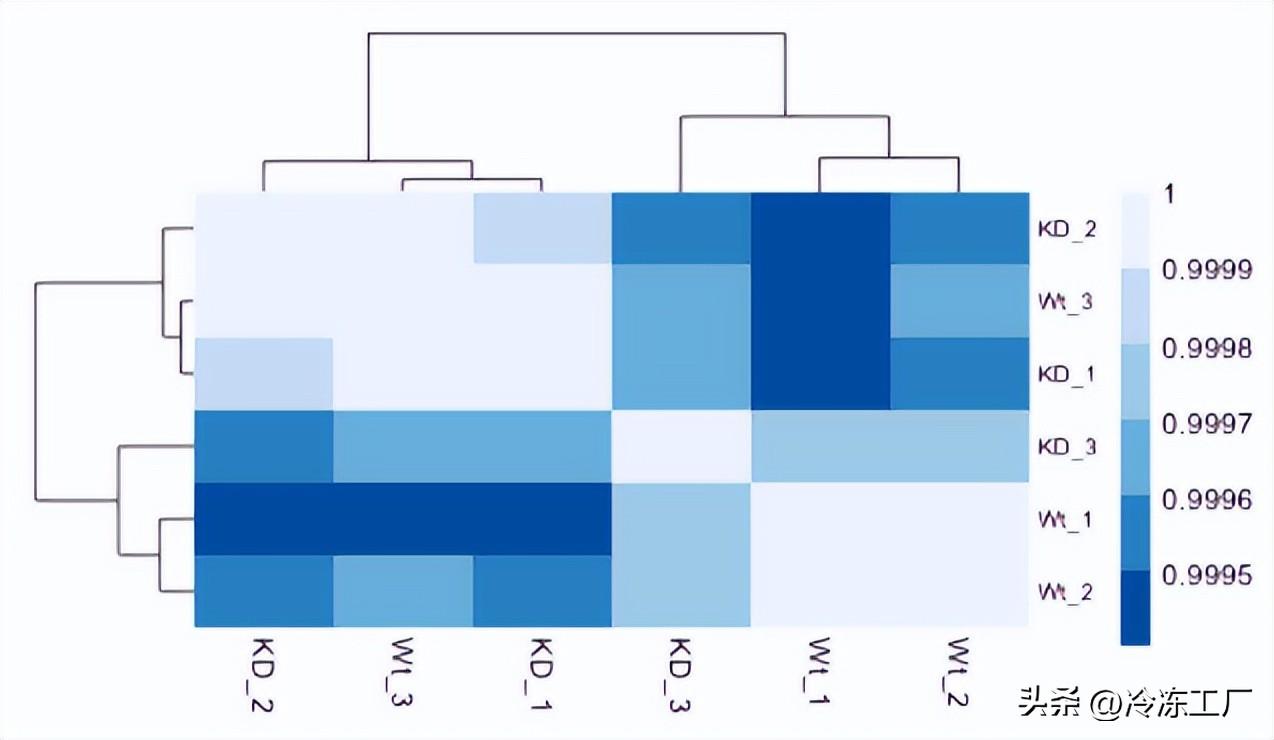
Hierarchical Clustering Heatmap
5. Mov10 QC
现在我们已经很好地理解了通常用于 RNA-seq 的 QC 步骤,让我们为 Mov10 数据集进行 QC。
5.1. 数据转换
为了促进 PCA 和层次聚类可视化方法的距离或聚类,我们需要通过对归一化计数应用 rlog 变换来调节均值的方差。
❝
归一化计数的 rlog 转换仅在该质量评估期间对于这些可视化方法是必需的。我们不会使用这些转换后的计数来确定差异表达。
❞
# rlog 转换
rld <- rlog(dds, blind=TRUE)
blind=TRUE 参数是为了确保 rlog() 函数不考虑我们的样本组——即以无偏见的方式进行转换。在执行质量评估时,包含此选项很重要。
rlog() 函数返回一个 DESeqTransform 对象,这是另一种特定于 DESeq 的对象。您不只是获得转换值矩阵的原因是因为用于计算 rlog 转换的所有参数(即大小因子)都存储在该对象中。我们使用此对象绘制 PCA 和层次聚类图以进行质量评估。
5.2. PCA
我们现在已准备好进行 QC 步骤,让我们从 PCA 开始吧!
DESeq2 有一个内置函数,可以在后台使用 ggplot2生成 PCA 图。这很棒,因为它使我们不必输入代码行,也不必摆弄不同的 ggplot2 层。此外,它直接将 rlog 对象作为输入,从而省去了我们从中提取相关信息的麻烦。
plotPCA() 需要两个参数作为输入:DESeqTransform 对象和 intgroup,即元数据中包含有关实验样本组信息列的名称。
# Plot PCA
plotPCA(rld, intgroup="sampletype")
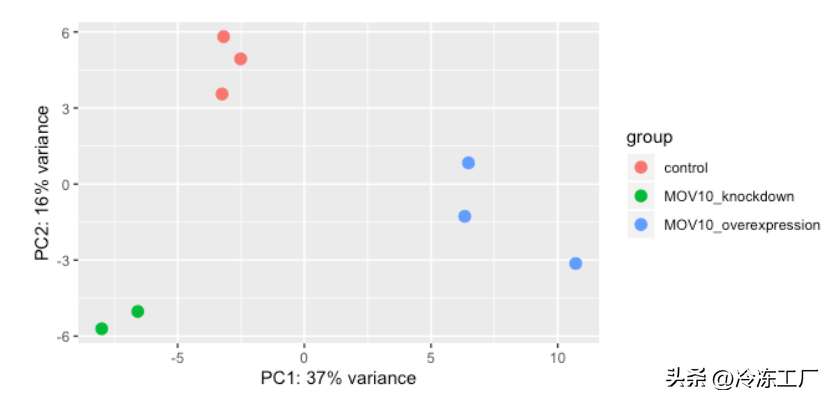
PCA
默认情况下,plotPCA()使用前 500 个最易变的基因。您可以通过添加 ntop= 参数并指定您希望函数考虑的基因数量来更改此设置。
plotPCA() 函数将只返回 PC1 和 PC2 的值。如果您想探索数据中的其他 PC,或者如果您想识别对 PC 贡献最大的基因,您可以使用 prcomp() 函数。例如,要绘制任何 PC,我们可以运行以下代码:
# Input is a matrix of log transformed values
rld <- rlog(dds, blind=T)
rld_mat <- assay(rld)
pca <- prcomp(t(rld_mat))
# Create data frame with metadata and PC3 and PC4 values for input to ggplot
df <- cbind(meta, pca$x)
ggplot(df) + geom_point(aes(x=PC3, y=PC4, color = sampletype))
5.3. Hierarchical Clustering
DESeq2中没有内置函数来绘制热图来显示所有样本之间的成对相关性和层次聚类信息;我们将使用 pheatmap 包中的 pheatmap() 函数。此函数不能使用 DESeqTransform 对象作为输入,但需要矩阵或数据框。因此,要做的第一件事是使用名为 assay() 的函数,从 rld 对象检索该信息,该函数将 DESeqTransform 对象中的数据转换为简单的二维数据结构。
# Extract the rlog matrix from the object
rld_mat <- assay(rld)
接下来,我们需要计算所有样本的成对相关值。我们可以使用 cor() 函数来做到这一点:
# Compute pairwise correlation values
rld_cor <- cor(rld_mat)
让我们看一下相关矩阵的列名和行名。
head(rld_cor)
head(meta)
您会注意到它们与我们在开始时使用的元数据数据框中为样本提供的名称相匹配。这很重要,因此我们可以使用下面的注释参数在顶部绘制一个色块。此块可轻松实现层次聚类的可视化。
# Load pheatmap package
library(pheatmap)
# Plot heatmap using the correlation matrix and the metadata object
pheatmap(rld_cor, annotation = meta)
当您使用 pheatmap() 进行绘图时,层次聚类信息用于将相似的样本放在一起,并且该信息由沿轴的树结构表示。注释参数接受一个数据框作为输入,在我们的例子中它是元数据框。
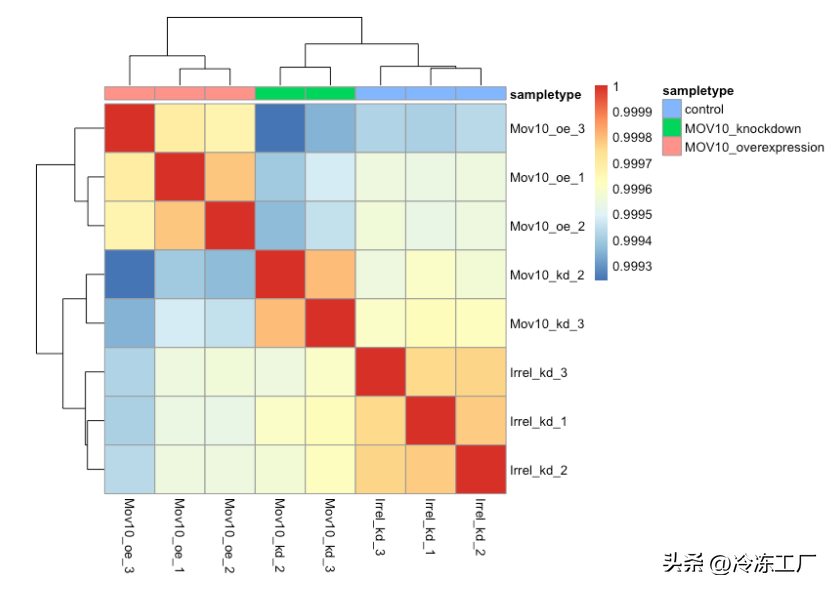
pheatmap
总体而言,我们观察到高相关性 (> 0.999),表明没有异常样本。此外,与 PCA 图类似,您会看到样本按样本组聚集在一起。总之,这些图向我们表明数据质量很好,我们有信心可以进行差异表达分析。





发表评论